JBrowse 2 provides considerable support for adding your own data. The first point about adding your own data is this: it doesn’t get uploaded to anywhere. The data you add always stays where it starts, there is no “uploading” of the data. This is an important point if you are worried about the privacy of your data.
JBrowse supports several common formats for genomics data, including GFF, VCF, BED, BigWig, BigBed, and BAM/CRAM. For a complete list of supported formats, see the JBrowse 2 documentation. For supported formats, the only caveat is that the reference sequence names should be names that the WormBase JBrowse will recognize. In this case, that means chromosome names like I, II, etc for C. elegans should be used, though JBrowse also understands aliases like chrI and chrII, which is what the UCSC Genome Browser uses.
As a first example of what can be done with your own data, we created a GFF file that has entries for polymorphisms that are found in the Hawaiian strain of C. elegans. We did this with a simple `grep` command, like
grep “\tVariation_project_Polymorphism\t” c_elegans.PRJNA13758.WS289.annotations.gff3.gz | grep CB4856 > hawaiian_variants.gff
To create a track for this data in the N2 JBrowse instance, there are two places I can find the “Add track” option. Both of these reside in the track selector, so if it’s not already open, click the big “open track selector” button in the linear genome view that appears when there are no tracks open,, or click the “open track selector” button in the upper left corner of the view. In the Available tracks panel, you can either click on the yellowish-orange circle with a plus sign or the “hamburger menu” at the upper left corner of the panel to get a menu with an “Add track” option.
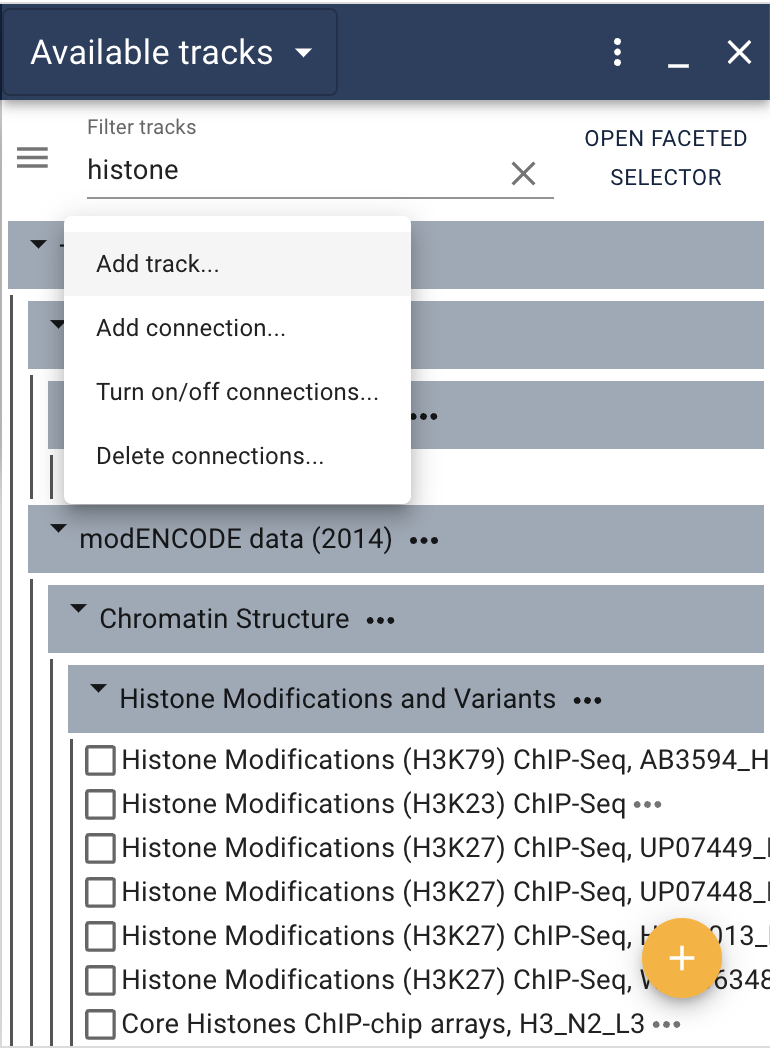
After selecting “Add track…” you are presented with a dialog to identify where the data is (JBrowse supports both local files and URLs for data). Select “File” to get a file browser to select the local file. In cases where there is an index file (like tabix-indexed GFF or VCF or indexed BAM files) you can also specify the location of the index (though if you don’t specify it, JBrowse will try to guess where the index is if it needs one).
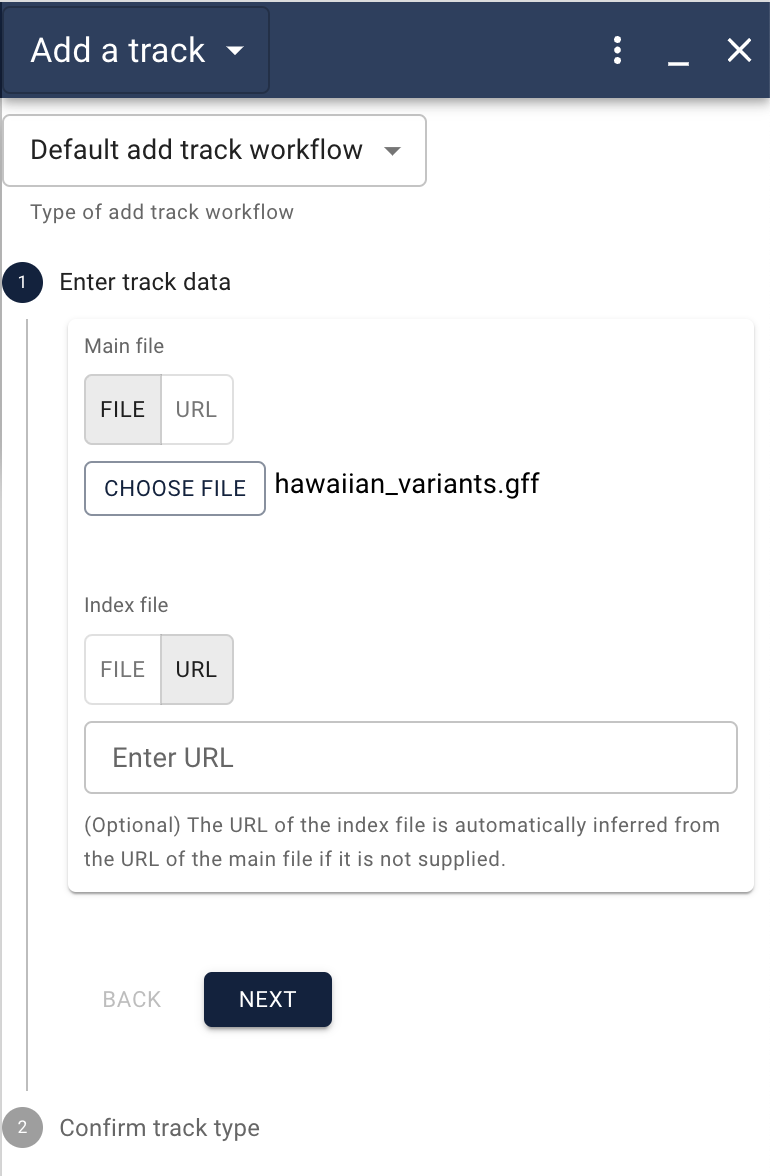
After clicking next, you’ll get a second dialog asking you to confirm information about the data the JBrowse has (usually correctly) assumed, though you will probably want to give the track a more descriptive name.
Clicking add then adds the track.
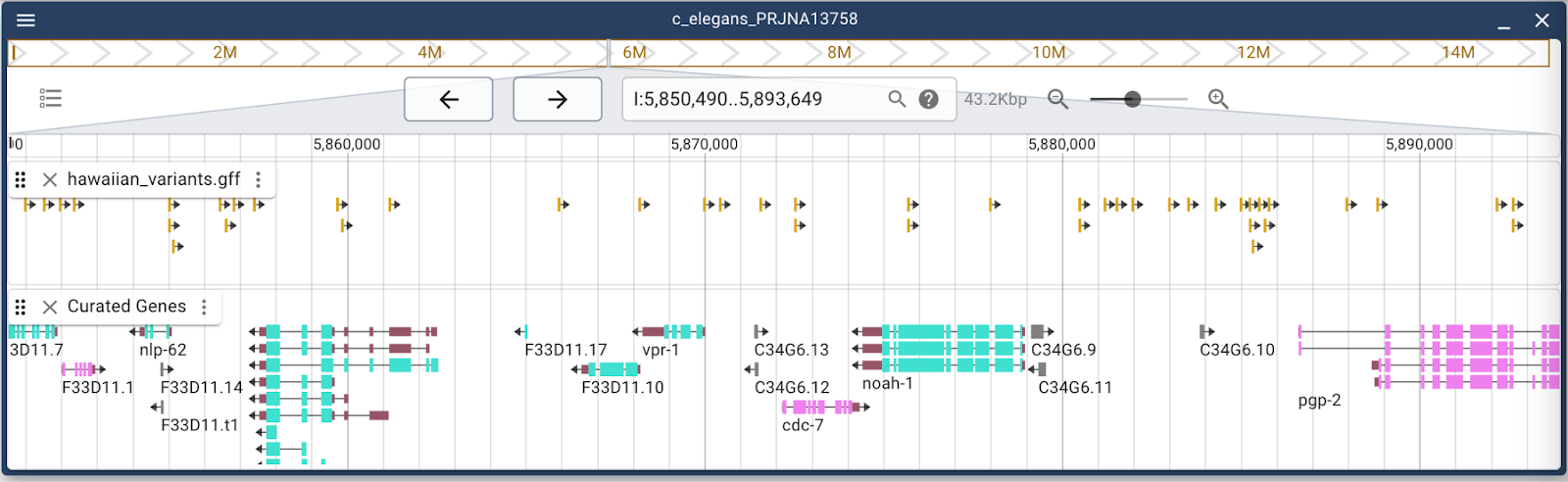
Note that now, this track has been added to the “Available tracks” panel under the “Session tracks” category. This is a reminder that this track will not persist through page closes or reloads. If you want to save this track, you should save your session. For more information on sessions, please see the “Sessions” section of JBrowse 2: Working with lists, bookmarks and sessions.
When you add your own data, you also have considerable flexibility to modify the way it looks. To change the details of the track, you can click the context menu button to the right of the track name in the “Available tracks” panel and select “Settings” from the menu.
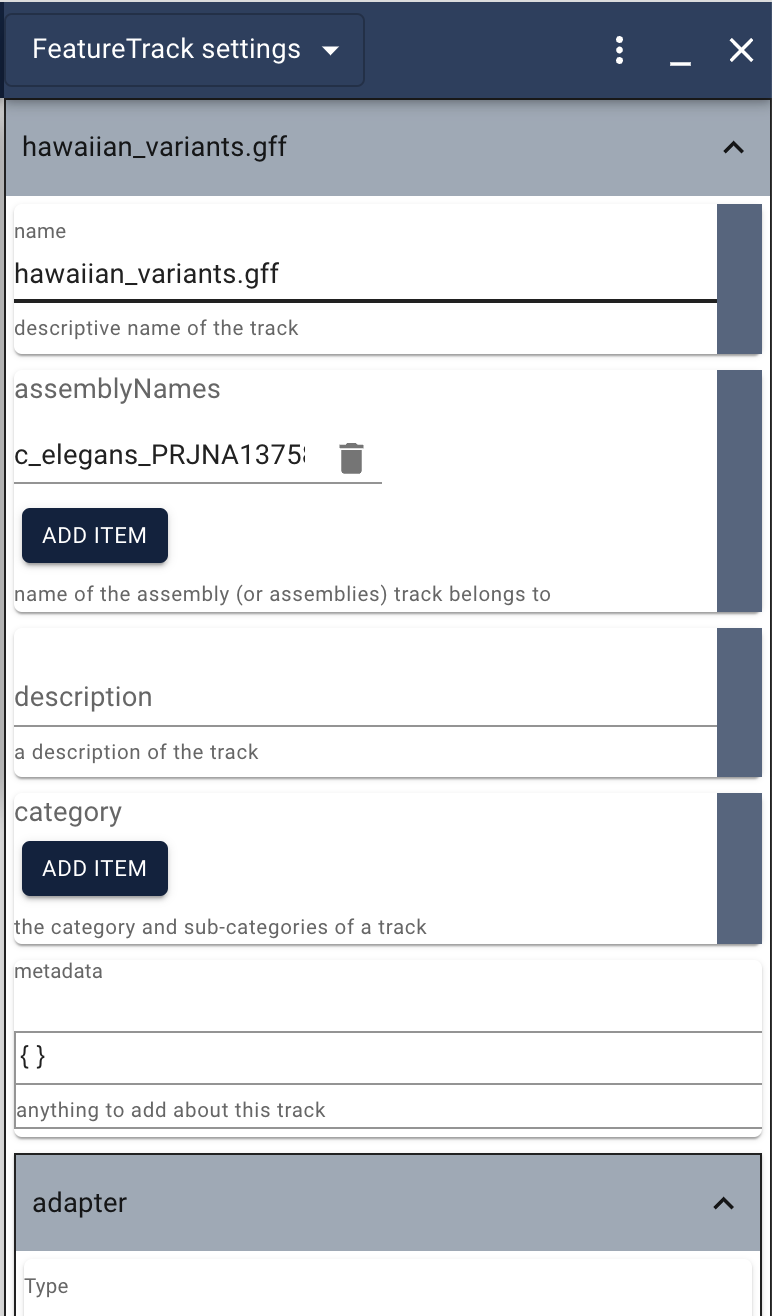
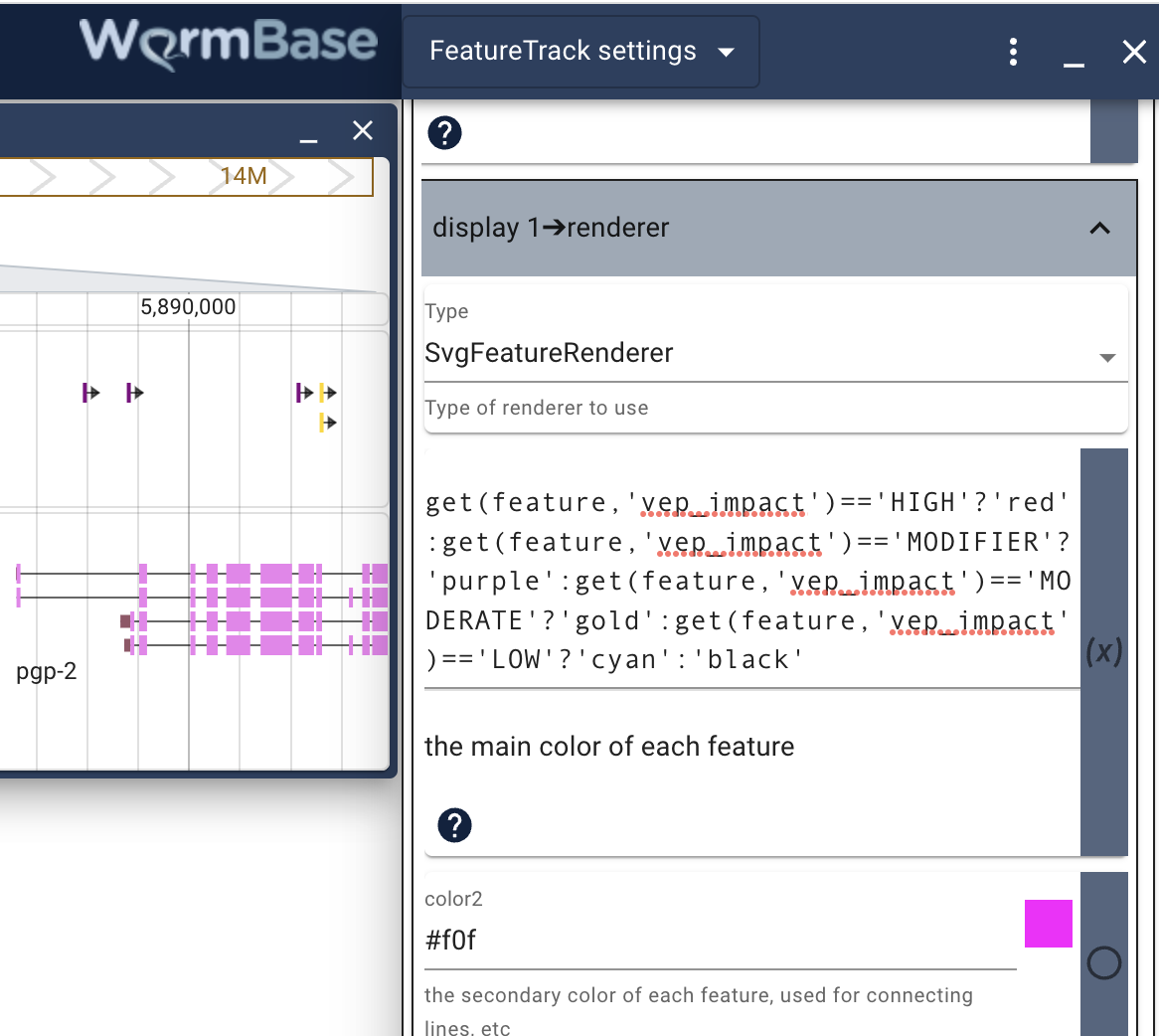
This is a very long list of options, so you’ll need to scroll down to see them all. You can change the name of the track, how the features are labeled and what the glyphs look like (size and color). In addition to changing things like the name of the color (from goldenrod to green for instance), you can also use a simple programming language called JEXL to change attributes of the features according to attributes of the feature. For example, if you wanted to change the color of the glyph using the same scheme that WormBase uses for their variant tracks (high impact red, medium gold, etc), you could add this code to the primary color (where the default is goldenrod). Note that to use JEXL where it’s not currently used you also have to check the button in the blue area to the right of the place where you want to use it.
get(feature,'vep_impact')=='HIGH'?'red':get(feature,'vep_impact')=='MODIFIER'?'purple':get(feature,'vep_impact')=='MODERATE'?'gold':get(feature,'vep_impact')=='LOW'?'cyan':'black'
Be careful with code copied and pasted from documents though: some times editors try to “help” and convert things like single quotes to “fancy” quotes, breaking the code.
Adding data from a URL
As you probably figured out from the Add track dialog, you can also load track data that is on the internet as well. If you have data or want to use data on a web server in a session track, the server must be configured to allow CORS (Cross Origin Resource Sharing) access. All of the data that WormBase uses for its JBrowse instances are shared with CORS allowed. Also, it is possible to add session tracks from services such as Google Drive and Dropbox, but requires some configuration on our end. If that is something you are interested in, please contact us through the WormBase help email address.
As an example of how to use web hosted data to add a session track we can use example data used for a JBrowse workshop at the Plant and Animal Genome conference in 2023. This is sequencing data comparing the Hawaiian strain to the N2 strain in the form of a BAM file. These data were generated by the C. elegans Natural Diversity Resource (http://elegansvariation.org/) and were used with their permission.
The BAM file and its .bai index file were placed in a public Amazon Web Services S3 bucket. The URL is this:
https://s3.amazonaws.com/wormbase-modencode/test/CeNDR/CB4854.bam
And the index file just has .bai appended to the name. This can be added with the same “Add track” menu. You can supply the URL of the index file, but if it is just the name of the original file with the index extension (as it is in this case), JBrowse will guess this location so it isn’t needed here.
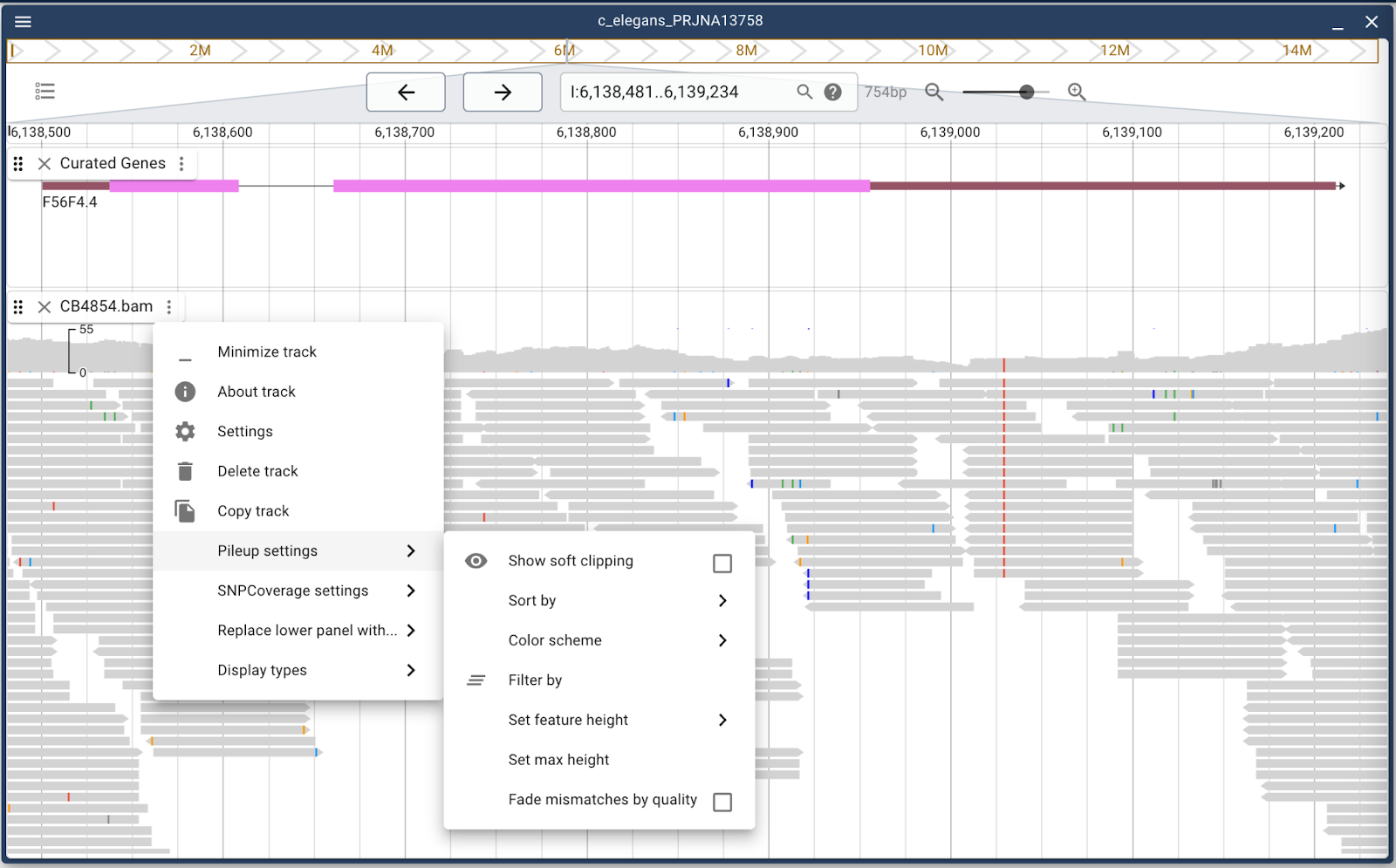
Alignment track settings
Since we’re here, we can also talk about dealing with alignment data like are found in BAM and CRAM files. The default display when a BAM file is loaded is to show both coverage and individual reads in gray with mismatches in other colors. There is considerable flexibility in how the track can display though. Either the individual reads or coverage plot can be turned off. Additionally, there are several read coloring schemes to make it easier to discern different aspects of the data. For example, mismatches can be set to “fade out” if the sequence quality isn’t good in that region. Also, alignments can be colored according to pairing, insert size, mapping quality or, in this example, per-base mapping quality:
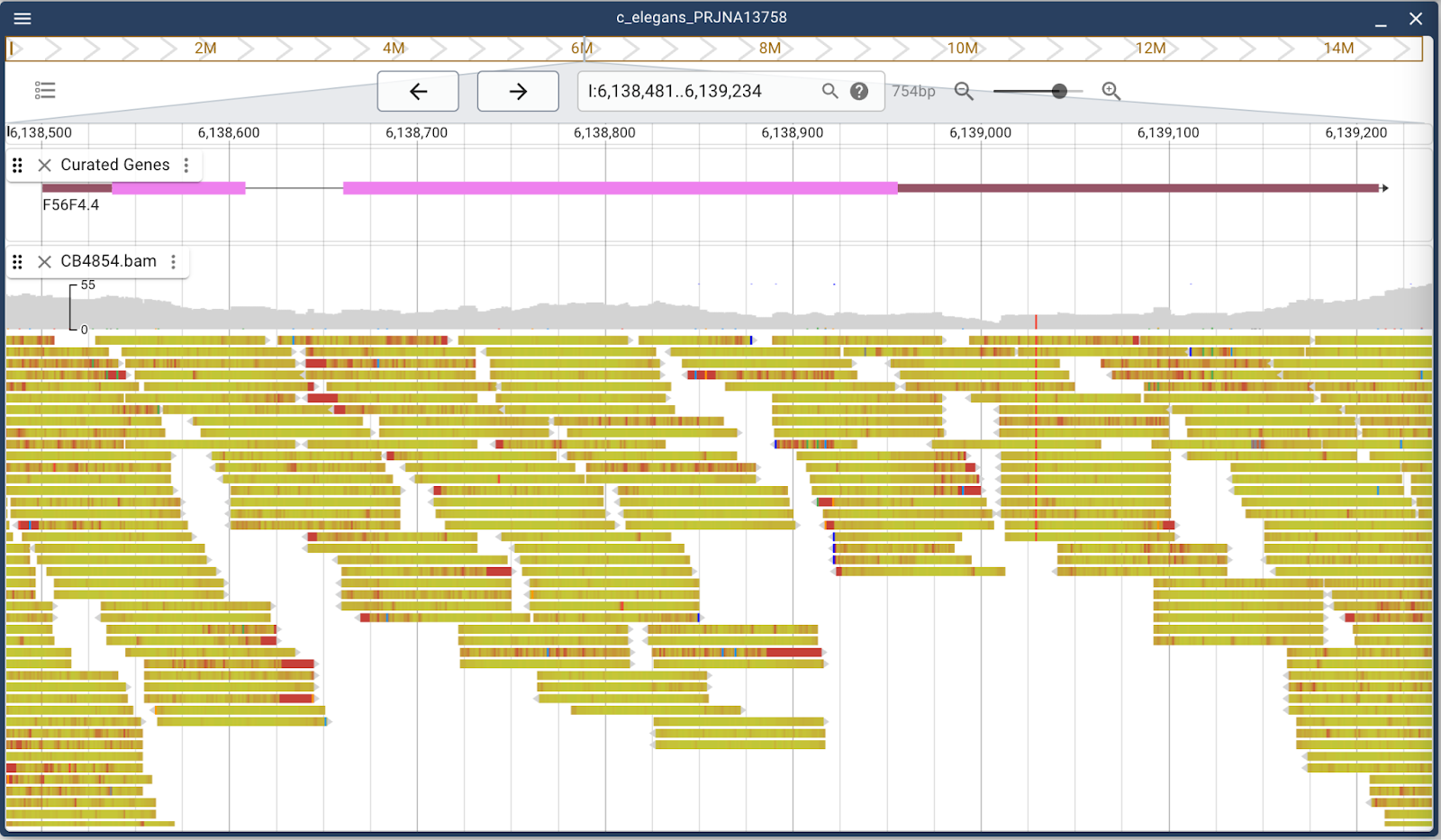
If you work with alignment data on a regular basis, it’s a good idea to explore the visualization options provided by JBrowse.
Leave a Reply